什么是先天性谷氨酰胺缺乏症?
先天性谷氨酰胺缺乏症是一种罕见的遗传代谢病。谷氨酰胺是人体内一种重要的氨基酸,参与多种生理过程,尤其在脑部能量供应和神经信号传递中扮演关键角色。当负责合成谷氨酰胺的关键基因发生突变,导致其功能严重受损时,身体就无法正常产生足够的谷氨酰胺。这种缺乏会直接影响大脑的正常发育和功能,从而引发一系列临床症状。
临床表现有哪些?
患有此病的婴儿通常在出生后不久就会出现严重问题。最突出的表现是严重的脑病,患儿可能出现意识障碍、反应迟钝、喂养困难。癫痫发作也很常见,形式多样且可能难以控制。部分患儿还会出现皮肤上的异常,表现为坏死性红斑,这是一种皮肤损伤。这些症状往往在新生儿期或婴儿早期就显现出来,进展迅速。需要强调的是,这些症状的出现顺序和严重程度因人而异。
这个病会遗传吗?
这种疾病的遗传方式是常染色体隐性遗传。这意味着,只有当一个人从父母双方那里各获得一个带有致病突变的基因拷贝时,才会患病。如果只从一方获得一个突变拷贝,另一方是正常的,这个人通常不会患病,但会成为携带者。对于一对健康的父母,如果他们恰好都是携带者,那么他们每次生育,孩子都有一定的概率(约25%)成为患者,也有概率成为健康的携带者或完全不受影响的孩子。了解这种遗传模式,有助于家庭成员理解疾病在家族中的传递可能性。
怎么确诊?
此类罕见遗传代谢病的诊断是一个综合过程。医生首先会基于临床表现,如严重的脑病、癫痫和皮肤特征,进行高度怀疑。随后,会进行一系列生化检查,可能包括血液和尿液中的氨基酸谱分析,以寻找谷氨酰胺水平异常或其他代谢物变化的线索。此类疾病通常通过基因检测辅助诊断,通过对相关基因进行测序分析,寻找是否存在已知的致病突变,从而从分子层面确认诊断。如有相关表现,建议就医由专科医生评估。
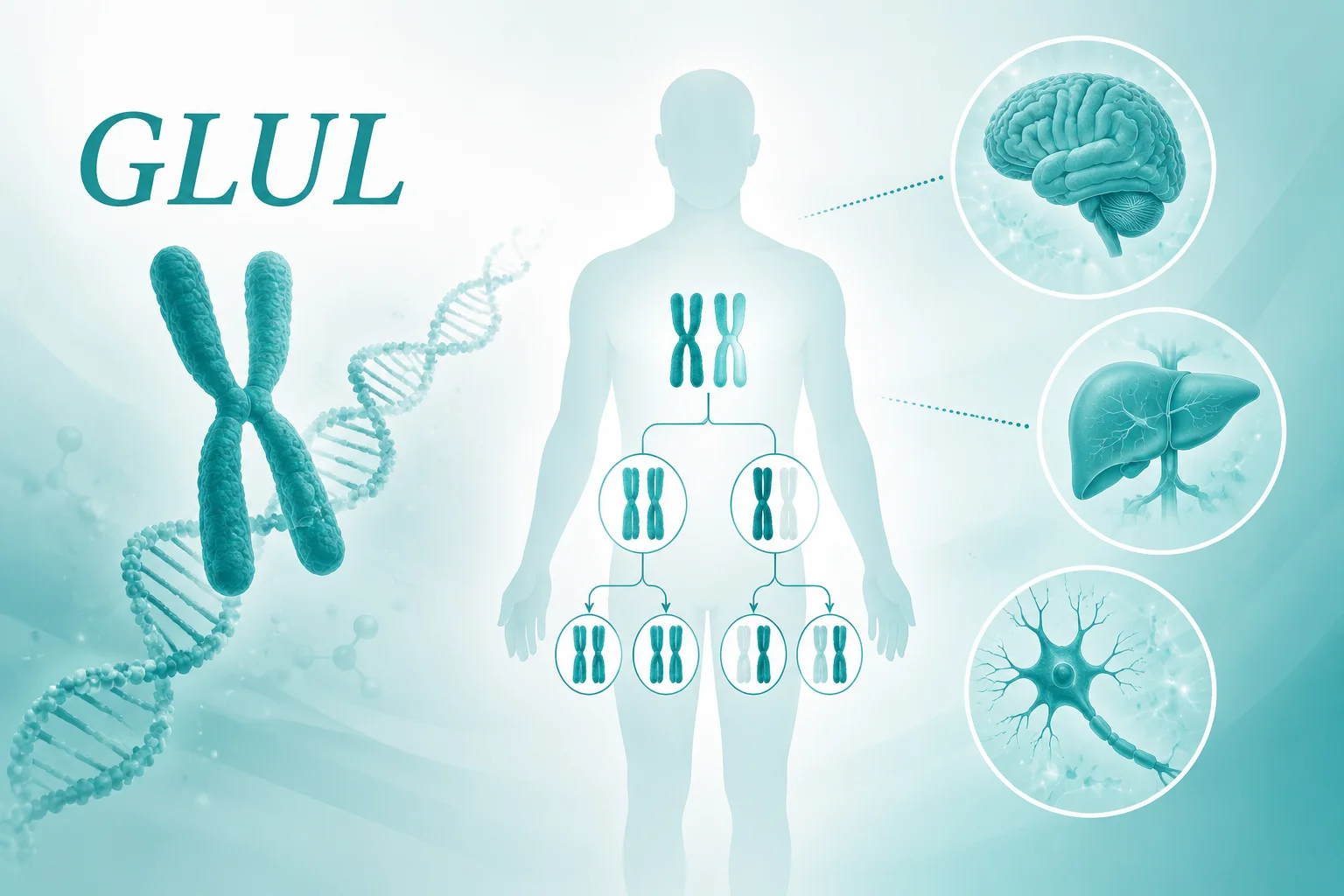
哪些基因相关?

在先天性谷氨酰胺缺乏症中,GLUL基因是关键之一。这个基因编码的蛋白质负责催化谷氨酰胺的合成。当GLUL基因发生致病性突变时,其编码的酶活性会显著下降或丧失,导致细胞,特别是脑细胞,无法获得足够的谷氨酰胺。基因检测的意义在于,它不仅能确认临床诊断,还能明确具体的基因突变类型,这对于理解疾病的分子基础、评估家族成员的携带状态以及未来可能的生育规划提供科学依据。
目前怎么治疗?预后如何?
目前,对于先天性谷氨酰胺缺乏症的治疗主要是支持性和对症治疗。目标是尽可能控制症状,改善患儿的生活质量。治疗措施可能包括使用抗癫痫药物来控制发作,提供特殊的营养支持,以及处理皮肤问题等。由于这是一种涉及基本代谢途径的先天缺陷,完全根治较为困难,预后很大程度上取决于诊断的及时性、症状的严重程度以及治疗干预的效果。医学研究仍在探索更有效的治疗方法。
能预防吗?家人需要筛查吗?
对于已经确诊的家庭,预防主要是指预防下一代再患同样疾病。基于明确的基因诊断,可以进行产前诊断。即在后续怀孕时,通过特定的技术手段对胎儿进行基因分析,了解其是否携带相同的致病突变。同时,对确诊患者的父母及其他家庭成员进行基因筛查(家系筛查),可以明确家族中的携带者情况,这能为家庭成员的生育决策提供遗传咨询信息。这些都属于在专业医疗指导下的可选方案。如有相关表现,建议就医由专科医生评估。